ASE Tutorials
- Introduction to ASE
- Getting Started
- Adsorption
- Transition States
- Error Estimation and Density of States
Adsorption on Surfaces
In the second exercise, you will be calculating the dissociative adsorption of N2 onto the your cluster and (111) surface. This will set you up for calculating the transition state in the third exercise. We will also calculate all the intermediates in the ammonia synthesis pathway (N*, NH*, NH2*, NH3*, H*)
Update (2016/02/20): Many of the clusters involving alloys or non-fcc metals underwent significant reconstruction during the relaxation. This leads to a highly asymmetric structure with many unique sites. These are completely new systems, so we don’t know their behavior a priori. Exploring all the unique sites would take much more time, so we have prepared all clusters with a fixed cuboctahedral shape. The lattice constants have been optimized with respect to this fixed structure. If your structure underwent significant reconstruction or distortion, you should use this for the adsorption and transition state calculations. Just remember to keep the metal atoms fixed in all cases, or else the system will become distorted again. Look for the #### FOR FIXED CLUSTERS ONLY #### comment in the scripts and make sure the lines following that are uncommented.
You may download the fixed structures using:
wget http://chemeng444.github.io/Fixed_Lattice_Clusters/METALS.traj
where METALS needs to be replaced with your system (e.g. PtRe).
Update (2016/02/23): Even if your cluster did not distort in the previous part, you may find that they distort once adsorbates are added on. In this case you can also use the fixed structures above. If you want, you can also study the effect of relaxing the metal atoms that are directly in contact with the adsorbates (instructions in the updated opt.py script). Remember that the exercises are only the minimum requirements for the project. A comparison between geometric and electronic effects is something that you can explore.
Update (2016/02/25): If you accidentally relaxed your cluster or if you find that your cluster distorts and after some optimization steps, here is a script to help you swap out the cluster with the fixed one, while keeping the adsorbate in the same place:
wget https://chemeng444.github.io/ASE/Adsorption/replace_with_fixed_cluster.py
to use this, run
python replace_with_fixed_cluster.py file.traj
and it will replace the cluster in file.traj with its fixed version.
Contents
Required Files
Obtain the required files by running:
on Sherlock:
cd $SCRATCH
wget http://chemeng444.github.io/ASE/Adsorption/exercise_2_sherlock.tar
tar -xvf exercise_2_sherlock.tar
or on CEES:
cd ~/$USER
wget http://chemeng444.github.io/ASE/Adsorption/exercise_2_cees.tar
tar -xvf exercise_2_cees.tar
This will create a folder called Exercise_2_Adsorption.
Gaseous Molecules
In this exercise you will be calculating the dissociative adsorption of N2 on your extended surface and your M13 surface from the previous exercise. The dissociative adsorption energy is defined as:
where N* refers to adsorbed N. We have Esurface from the previous exercise, so we will need to calculate both Esurface + 2N* and EN2 in this exercise.
In the N2_gas subfolder, find the run_N2.py script. This is a typical script for calculating gas phase species: the optimized geometry is determined, then the electronic energy, as well as the vibrational modes are computed and used to determine the free energy. Run the script and check that the vibrational modes are reasonable. Ideally, one large vibrational frequency corresponding to the N-N stretching should be observed.
Update (2016/02/20): There was a typo in run_N2.py where the vacuum was mistakenly doubled in all directions. Either download the script again or change line 17 to:
atoms.center(10.0)
Adsorption Sites
Take a look here if you need a reminder on how to add atoms using ase-gui. We will describe how to add atoms within the ASE script below.
Enter the Adsorption subfolder.
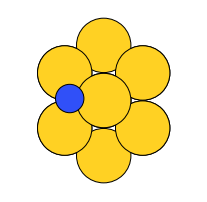
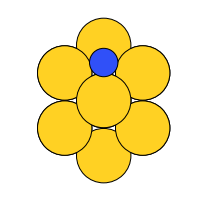
There are four possible adsorption sites on fcc (111) surfaces that an adsorbate can bind to: the fcc, hcp, ontop, and bridge sites. For a bcc (110), there are three sites: hollow, ontop, and bridge. For the M13 clusters, there are four sites: 1-fold, 2-fold, 3-fold, and 4-fold coordinated sites. These are illustrated below:
| M13 | 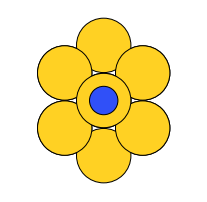 on-top |
 two-fold |
 three-fold |
 four-fold |
| fcc (111) | 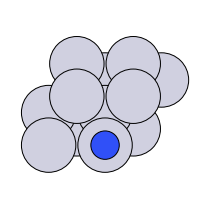 on-top |
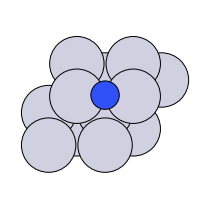 bridge |

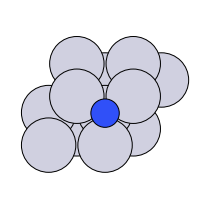 fcc, hcp |
|
| bcc (110) | 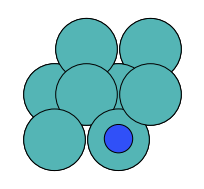 on-top |
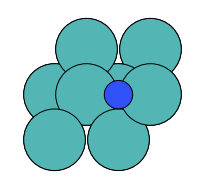 bridge |
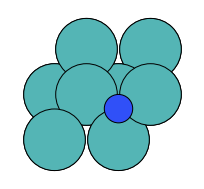 hollow |
|
If you are working with an alloy system, it is possible that the structure has distorted significantly. In that case, just identify the unique adsorption sites for your system.
In the setup_ads.py script, an optimized structure is read, and then the adsorbate atoms are added manually. This script can be executed in the login node using python setup_ads.py. The add_adsorbate() function is used to add adsorbates:
add_adsorbate(slab, 'N', 1.5, (3, 1.7))
add_adsorbate(slab, 'N', 1.5, (1.5, 0.86))
Here add_adsorbate(slab, element, z, (x,y) is used, where slab is the loaded trajectory file, z is the vertical height above the surface, and (x,y) are the x and y coordinates.
Edit the script to add the adsorbates in the sites you need.
Alternatively, for extended surfaces, one can first use the fcc111 (bcc110 if you are calculating Mo) function from the ase.lattice.surface module to set up the surface. Then, special keywords in the add_adsorbate() function can be directly used to add adsorbates to the special sites, e.g.
atoms = fcc111('Pt', a = 3.989, size = (2,2,4), vacuum = 7.0)
add_adsorbate(atoms, 'N', 1.5, 'fcc')
add_adsorbate(atoms, 'N', 1.5, 'hcp')
This can only be done if the surface has just been created, and not if it is being read from a saved .traj file. Furthermore, the function can only add one adsorbate on one type of site. For example, you cannot add two N adsorbates onto two different bridge sites. It will add them both onto the same bridge site. More information can be found here. These lines can be added at the beginning of the optimization script. If you are already reading in a trajectory using atoms = io.read('trajfile.traj'), then this will just overwrite atoms with a new surface, so be careful!
One could also use the ASE graphical user interface ase-gui to add an adsorbate or molecule as well. Use ase-gui <file>.traj to open the trajectory file. Then simply click the atom above where the adsorbate will sit, and click Ctrl + A, then specify the adsorbate and the vertical distance above the site. You can also hold Ctrl to select multiple atoms and add an adsorbate, which will be at the center of all the selected atoms.
Dissociative adsorption
We will focus on the dissociative adsorption mechanism for the first bond-breaking step of N2, where the N2 molecule separates as two N* adsorbed onto the surface. In this case you need to check all unique neighboring sites for the two N* atoms.
You will be making comparisons between the M13 cluster and the surface for the 2N* state and the dissociation barrier.
For the rest of the ammonia synthesis pathway, you are only required to perform calculations on the M13 cluster. If time allows, you are welcome to do more calculations.
Organize your directories in the following way:
../Adsorption/2N_surface/
../Adsorption/2N_cluster/
../Adsorption/N/
../Adsorption/N/ontop
../Adsorption/N/bridge
...
../Adsorption/NH3/
Once you have all your structures set up, it is time to run the structural optimization. The opt.py script should be submitted within each subdirectory for each adsorption site. This way the output files will be written to their respective subdirectories.
Note: You will need to explore all possible adsorption sites to find the ones with the lowest energy, as those will be the most stable configurations. For the 2N* calculation, you will need to focus on neighboring sites. However, what constitutes a “neighboring” site might depend on your system. You might find that when the two N* atoms are close to each other, that they combine to form an adsorbed N2* during the optimization. This just means that your surface may be so reactive that it can stabilize the N2 molecule without relying on the N-N bond being broken. In that case you will have to look for sites slightly further apart.
Requirement: Complete structural optimizations for the following adsorbates:
- Calculate 2N* adsorption on all possible sites on the M13 cluster and the extended surface.
- Calculate the reaction intermediates (from N* through NH3*) on all possible adsorption sites only for the M13 cluster.
- There may be a lot of possible configurations, so we recommend that you set up all possible ones and submit them at the same time.
Batch Submission
To save you some time, you can try using the following script for batch submitting the optimization jobs:
export CURDIR=$PWD #save current directory
for ads in 2N_surface 2N_cluster N NH NH2 NH3 H #loop through first directory
do
for site in $ads/*/ #loop through ALL subdirectories of ads folder
do #you can also specify individually or use wildcards
# cp opt.py $site #uncomment to copy opt.py to every subdir
cd $site
if [[ $hostname == *"sherlock"* ]]
then
sbatch --job-name=$PWD opt.py #sbatch if sherlock
else
qsub opt.py #qsub if cees
fi
cd $CURDIR #go back to the original directory and continue loop
done
done
the script simply loops through the folders 2N_surface 2N_cluster N NH NH2 NH3 H, and all of their subdirectories and executes the submission sbatch or qstat command.
Get the script here
wget http://chemeng444.github.io/ASE/Adsorption/batchsub.sh
and execute from the directory where N NH ... NH3 ... H are using
/usr/bin/env bash batchsub.sh
Next: move on to Transition States to learn about how to determine transition states and barriers.